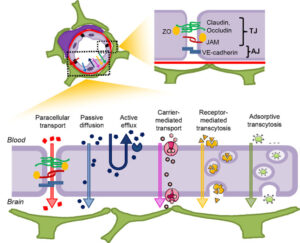
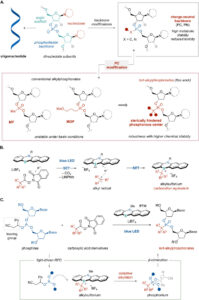
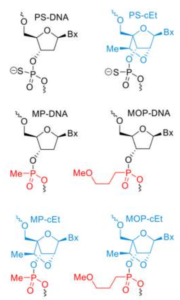
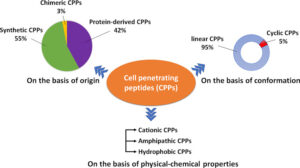
Peptides as Pharmacological Carriers to the Brain: Promises, Shortcomings and Challenges 2 min read Peptides Peptides as Pharmacological Carriers to the Brain: Promises, Shortcomings and Challenges April 21, 2023 Central nervous system (CNS) diseases are among the most difficult to treat, mainly because the vast majority...Read More